1. RNA methylation regulates transcription and chromatin dynamics

m6A of chromosome-associated regulatory RNA regulates chromatin state and transcription
m6A methylation plays a crucial role in mouse embryonic development. Knockout of the m6A methyltransferase METTL3 results in embryonic lethality in mice, a phenotype closely resembling the effect of knocking out the nuclear m6A reader protein YTHDC1, indicating an essential nuclear function for m6A. Through analysis of m6A MeRIP-seq data for chromatin-associated RNAs, we found that these RNAs, particularly non-coding RNAs, undergo m6A methylation, with significantly reduced m6A levels upon METTL3 knockout. These non-coding RNAs are highly enriched at genomic regulatory elements, leading us to define them as chromatin-associated regulatory RNAs (carRNAs), which include promoter-associated RNAs (paRNAs), enhancer RNAs (eRNAs), and transposon-derived RNAs (repeat RNAs). Mechanistic studies reveal that m6A-methylated carRNAs are recognized by YTHDC1, which recruits the nuclear exosome targeting (NEXT) complex to mediate their degradation, resulting in chromatin silencing and repression of downstream gene transcription. Furthermore, by employing the dCas13b system, we confirmed the causal role of this regulatory pathway and its indispensable function in embryonic development (Science 2020).
RNA m5C oxidation by TET2 regulates chromatin state and leukaemogenesis
Mutation of tet methylcytosine dioxygenase 2 (encoded by TET2) drives myeloid malignancy initiation and progression. TET2 deficiency is known to cause a globally opened chromatin state and activation of genes contributing to aberrant haematopoietic stem cell self-renewal. However, the open chromatin observed in TET2-deficient mouse embryonic stem cells, leukaemic cells and haematopoietic stem and progenitor cells is inconsistent with the designated role of DNA 5-methylcytosine oxidation of TET2. Here we show that chromatin-associated retrotransposon RNA 5-methylcytosine (m5C) can be recognized by the methyl-CpG-binding-domain protein MBD6, which guides deubiquitination of nearby monoubiquitinated Lys119 of histone H2A (H2AK119ub) to promote an open chromatin state. TET2 oxidizes m5C and antagonizes this MBD6-dependent H2AK119ub deubiquitination. TET2 depletion thereby leads to globally decreased H2AK119ub, more open chromatin and increased transcription in stem cells. TET2-mutant human leukaemia becomes dependent on this gene activation pathway, with MBD6 depletion selectively blocking proliferation of TET2-mutant leukaemic cells and largely reversing the haematopoiesis defects caused by Tet2 loss in mouse models. Together, our findings reveal a chromatin regulation pathway by TET2 through retrotransposon RNA m5C oxidation and identify the downstream MBD6 protein as a feasible target for developing therapies specific against TET2 mutant malignancies (Nature 2024).
2. RNA methylation regulators determine their functional specificity
RBFOX2 recognizes paRNA m6A to suppress transcription and block myeloid leukaemia differentiation
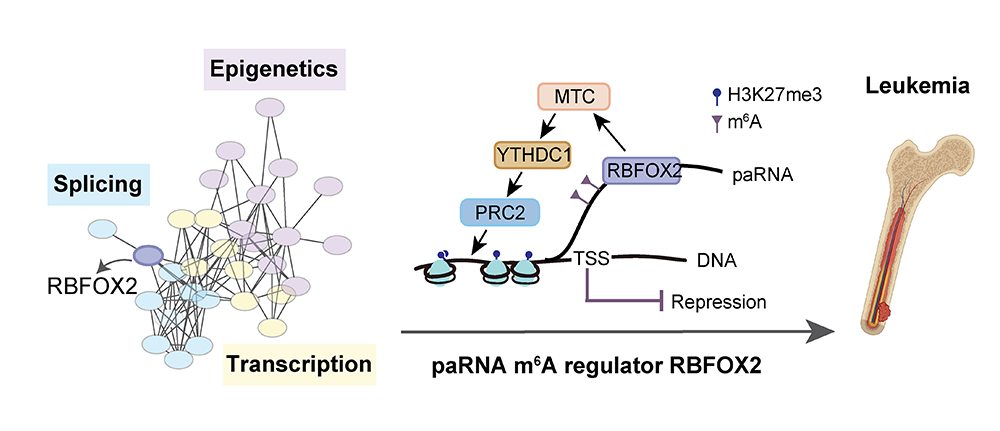 Chromatin binds various non-coding RNAs with regulatory functions, which are
modified to
varying degrees by m6A methylation. However, there is still a lack of
understanding about the regulatory proteins that can specifically recognize these non-coding
RNAs and their chemical modifications, thereby mediating changes in chromatin state and gene
transcription activity. To address this gap, we conducted the first integrated analysis of
large-scale protein binding data across the whole transcriptome and genome, as well as
m6A methylation data of chromatin-associated RNAs, systematically identifying
potential m6A regulatory proteins on chromatin-associated RNAs, including the
classic alternative splicing factor RBFOX2. We discovered that RBFOX2, independently of its
splicing function, specifically recognizes m6A-methylated chromatin-associated
RNAs, particularly promoter-associated RNAs (paRNAs). RBFOX2 then recruits the
m6A methyltransferase complex (MTC) to gene promoter regions via RBM15. At the
same time, RBFOX2 interacts with YTHDC1 through RBM15, recruiting the polycomb repressive
complex 2 (PRC2) to the promoter region, thereby initiating chromatin silencing and
downstream gene transcriptional repression. Upon RBFOX2 knockdown, PRC2 binding to chromatin
is significantly reduced, and this reduction occurs only at sites where RBM15 or YTHDC1 is
bound, confirming that PRC2 recruitment to RBFOX2 binding sites requires the assistance of
RBM15 and YTHDC1. Further studies revealed that in a patient-derived tumor
xenotransplantation (PDX) model, knockdown of RBFOX2 not only significantly promoted the
differentiation of acute leukemia cells but also markedly extended mouse survival. This
effect is achieved by regulating the TGF-β signaling pathway through the
m6A/RBM15/YTHDC1/PRC2 molecular mechanism (Nature Cell Biology,
2023).
Chromatin binds various non-coding RNAs with regulatory functions, which are
modified to
varying degrees by m6A methylation. However, there is still a lack of
understanding about the regulatory proteins that can specifically recognize these non-coding
RNAs and their chemical modifications, thereby mediating changes in chromatin state and gene
transcription activity. To address this gap, we conducted the first integrated analysis of
large-scale protein binding data across the whole transcriptome and genome, as well as
m6A methylation data of chromatin-associated RNAs, systematically identifying
potential m6A regulatory proteins on chromatin-associated RNAs, including the
classic alternative splicing factor RBFOX2. We discovered that RBFOX2, independently of its
splicing function, specifically recognizes m6A-methylated chromatin-associated
RNAs, particularly promoter-associated RNAs (paRNAs). RBFOX2 then recruits the
m6A methyltransferase complex (MTC) to gene promoter regions via RBM15. At the
same time, RBFOX2 interacts with YTHDC1 through RBM15, recruiting the polycomb repressive
complex 2 (PRC2) to the promoter region, thereby initiating chromatin silencing and
downstream gene transcriptional repression. Upon RBFOX2 knockdown, PRC2 binding to chromatin
is significantly reduced, and this reduction occurs only at sites where RBM15 or YTHDC1 is
bound, confirming that PRC2 recruitment to RBFOX2 binding sites requires the assistance of
RBM15 and YTHDC1. Further studies revealed that in a patient-derived tumor
xenotransplantation (PDX) model, knockdown of RBFOX2 not only significantly promoted the
differentiation of acute leukemia cells but also markedly extended mouse survival. This
effect is achieved by regulating the TGF-β signaling pathway through the
m6A/RBM15/YTHDC1/PRC2 molecular mechanism (Nature Cell Biology,
2023).
Exon architecture controls mRNA m6A suppression and gene expression
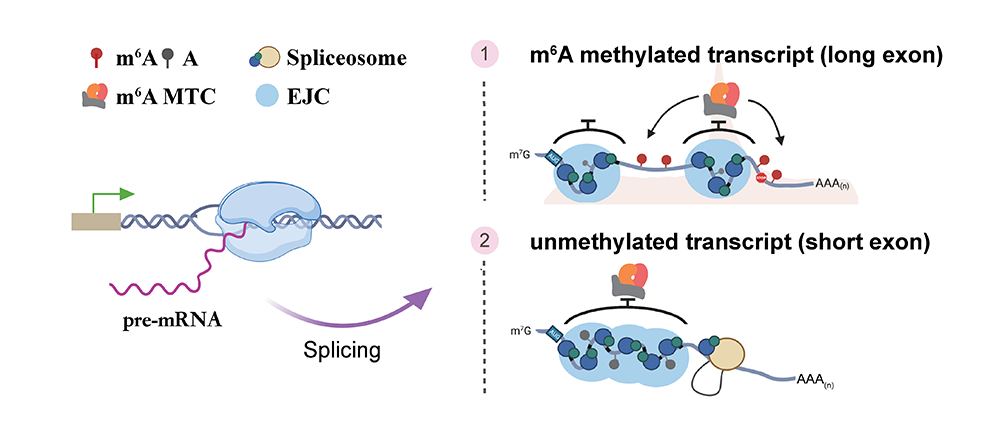 In mammalian cells, m6A methylation exhibits significant regional
selectivity
across the transcriptome, with high enrichment near unusually long exons and stop codons.
This selectivity is crucial for m6A-mediated gene regulation. However, our
understanding of the underlying mechanisms remains limited. Using a high-throughput parallel
reporter assay to measure m6A, the applicant and collaborators found that
alternative splicing spatially hinders the m6A methyltransferase complex from
accessing short exons, thereby inhibiting their m6A methylation. This process
determines several key features of the transcriptome-wide specific distribution of
m6A methylation. Consistently, when the exon junction complex (EJC) is knocked
out, m6A levels rise significantly, particularly at sites on short exons and
regions far from stop codons. Further computational analyses show that this structural
characteristic of exons is highly conserved throughout vertebrate evolution, influencing
both the distribution of m6A methylation and YTHDF2-mediated mRNA stability
(Science, 2023).
In mammalian cells, m6A methylation exhibits significant regional
selectivity
across the transcriptome, with high enrichment near unusually long exons and stop codons.
This selectivity is crucial for m6A-mediated gene regulation. However, our
understanding of the underlying mechanisms remains limited. Using a high-throughput parallel
reporter assay to measure m6A, the applicant and collaborators found that
alternative splicing spatially hinders the m6A methyltransferase complex from
accessing short exons, thereby inhibiting their m6A methylation. This process
determines several key features of the transcriptome-wide specific distribution of
m6A methylation. Consistently, when the exon junction complex (EJC) is knocked
out, m6A levels rise significantly, particularly at sites on short exons and
regions far from stop codons. Further computational analyses show that this structural
characteristic of exons is highly conserved throughout vertebrate evolution, influencing
both the distribution of m6A methylation and YTHDF2-mediated mRNA stability
(Science, 2023).
3. RNA methylation impacts human diseases, including cancer and neurodegenerative disorders
m6A in cancer immunotherapy
In the tumor microenvironment, immune suppression led by myeloid-derived suppressor cells (MDSCs) is a major barrier to the effectiveness of radiotherapy and combined radiotherapy-immunotherapy, closely linked to poor prognosis in cancer patients. The applicant and collaborators discovered that radiotherapy rapidly induces the upregulation of the m6A-binding protein YTHDF2 in MDSCs through NF-κB activation. This upregulation promotes the degradation of m6A-methylated mRNAs of NF-κB negative regulators, forming a positive feedback loop that further activates the NF-κB signaling pathway. This process significantly enhances MDSC differentiation, infiltration, and immunosuppressive function, reducing the effectiveness of radiotherapy. Further research showed that targeting and inhibiting YTHDF2 markedly improves the response rates to existing radiotherapy and combined radiotherapy-immunotherapy, offering a powerful and practical strategy to address immune suppression in tumor radiotherapy (Cancer Cell, 2023).
m6A in neurodegenerative diseases
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease that typically manifests in middle age, leading to muscle paralysis, with no effective treatment currently available. ALS shares similar pathology and genetics with frontotemporal dementia (FTD), often caused by abnormal expansion of the GGGGCC repeat sequence in the non-coding region of the C9ORF72 gene, accompanied by RNA metabolism abnormalities, though the underlying mechanisms remain unclear. We found that a significant reduction in m6A levels on mRNA in patient neurons is a key factor in this RNA metabolism abnormality. By restoring m6A levels, the expression of GGGGCC repeat RNA in patient neurons was effectively reduced, nuclear RNA aggregates decreased, expression of endogenous dipeptide repeat proteins reduced, and neurons resistance to excitotoxicity was enhanced, ultimately improving the survival of patient neurons (Nature Neuroscience, 2023).